柯俊良團隊2024研究成果|不管對第三代標靶藥抗藥與否,小孢子靈芝免疫調節蛋白質GMI都能抑制EGFR雙突變肺腺癌細胞透過「整合素」另謀生路

- 詳細內容
- 分類:中山醫學大學 柯俊良教授
- 建立於 2025-03-07, 週五 15:34
- 點擊數:2411
對於一再突變,甚至是一再對EGFR標靶藥出現抗藥性的突變型EGFR肺腺癌,只能換個標靶姿勢再治一次,還是走回傳統化療的老路?中山醫學大學醫學研究所柯俊良教授率其團隊成員辛翌綸博士、康于庭博士(台中榮民總醫院博士後研究員)等發表在2024年12月號《Environmental Toxicology》的研究讓我們看到了另一種因應的可能性,那就是既能顧前(清除EGFR)也能顧後(阻止癌細胞透過「整合素」另謀生路)的小孢子靈芝免疫調節蛋白質GMI(Ganoderma microsporum immunomodulatory protein)。
文/吳亭瑤

對前兩代標靶藥有抗藥性的
EGFR雙突變的肺腺癌細胞
簡稱EGFR的「表皮生長因子受體」是眾多位於細胞表面的接受器之一,它可以接收多種外來信號,活化細胞內的相關分子來驅動細胞增生。正常細胞對EGFR的表現量和活性會嚴格調控,可是有些癌細胞會卻把EGFR當成利器,將其大量表現(即所謂的EGFR陽性)加快增生速度;甚或發展出突變的EGFR,毋須等待外來信號就能自行運作,讓細胞長得更快更多也更具侵襲性。
EGFR最常見的突變是L858R,意指EGFR這個蛋白的第858個胺基酸由亮胺酸(L)突變為精胺酸(R),其具體位置在EGFR位於細胞內部另一端的酪胺酸激酶區域。第一代標靶藥(吉非替尼/艾瑞莎、厄洛替泥/得舒緩)和第二代標靶藥(阿法替尼/吉泰瑞)均是專門針對這種突變而設計,藉由與酪胺酸激酶的活性位點結合,阻斷EGFR的訊息傳遞,抑制細胞增生。
然而經過一段時間治療之後EGFR通常會再次發生T790M突變,也就是第790個胺基酸從原本的蘇胺酸(T)變成了甲硫胺酸(M),導致前兩代EGFR標靶藥無法與酪胺酸激酶的活性位點順利結合而失去效用。本研究使用的人類肺腺癌細胞H1975就具有EGFR雙突變(L858R/T790M)的特性。
(1) GMI可抑制腫瘤生長
把H1975細胞注射到裸鼠(免疫有缺陷的小鼠)右側腹皮下組織,一週後再以口服方式每天給予160 μg GMI或生理食鹽水,結果GMI組的腫瘤不只長得慢、體積小,腫瘤組織裡具有活性的癌細胞也比較少【圖1】。這說明GMI是一個可以經由胃腸道發揮抗腫瘤作用的活性成分,而且可以在免疫不彰的體內環境下獨力抑制H1975細胞增生。
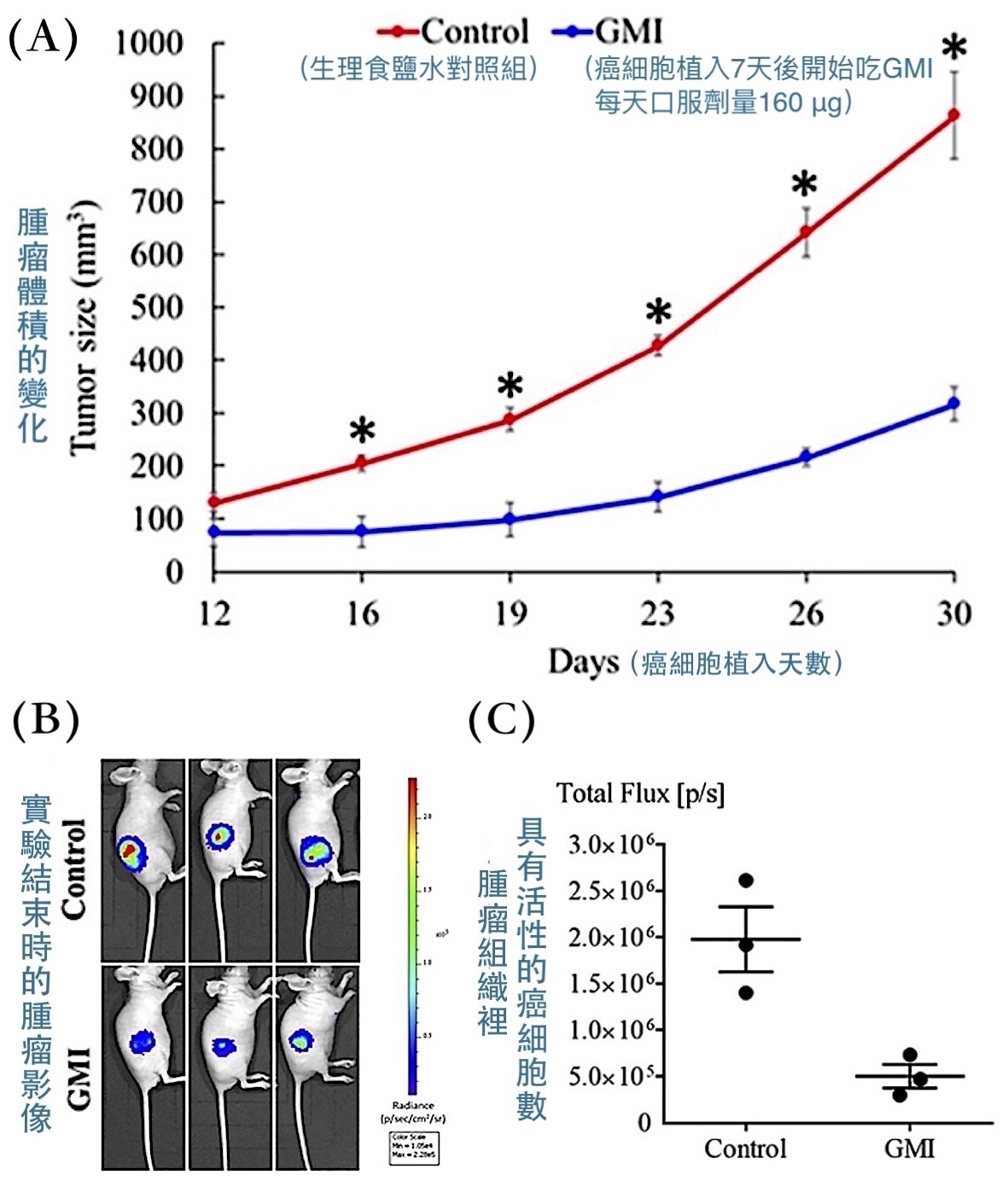
【圖1】GMI對裸鼠體內的H1975細胞有抑制腫瘤生長的作用
(2) GMI可降低癌細胞的遷移能力
體外模擬細胞遷移的實驗則顯示,H1975細胞的遷移能力【圖2-①】會在轉化生長因子TGF-β誘導下變得更強【圖2-③】,不過只要同時有GMI(0.3 μM)存在,不管有【圖2-④】或沒有【圖2-②】TGF-β誘導,癌細胞從兩旁移至中間無細胞區的數量均會下降,顯示GMI有抑制H1975細胞遷移的作用。

【圖2】GMI可在TGF-β誘導細胞遷移的環境下抑制H1975細胞遷移
對第三代標靶藥有抗藥性的
EGFR雙突變肺腺癌細胞
學名為奧希替尼(osimertinib)、商品名為泰格莎(Tagrisso)的小分子化合物,是專門設計來對付EGFR雙突變(L858R/T790M)癌細胞的第三代標靶藥。與前兩代標靶藥類似,奧希替尼也是透過與EGFR的酪胺酸激酶區域結合(但結合位點跟前兩代不同)來抑制癌細胞增生,而且也同樣難以避免幾個月後EGFR再次突變導致藥物失效的問題。
(1) GMI可降低癌細胞的存活率
為了了解GMI是否也能對付這樣的癌細胞,本研究利用H1975細胞建立了「EGFR雙突變但對奧希替尼有抗藥性」的人類肺腺癌細胞H1975/TR,結果發現,H1975/TR的存活率會隨GMI的使用濃度與作用時間而降低【圖3】,這說明GMI對H1975/TR細胞具有一定的殺傷力,也暗示GMI的作用機制有別於現有的EGFR標靶藥。
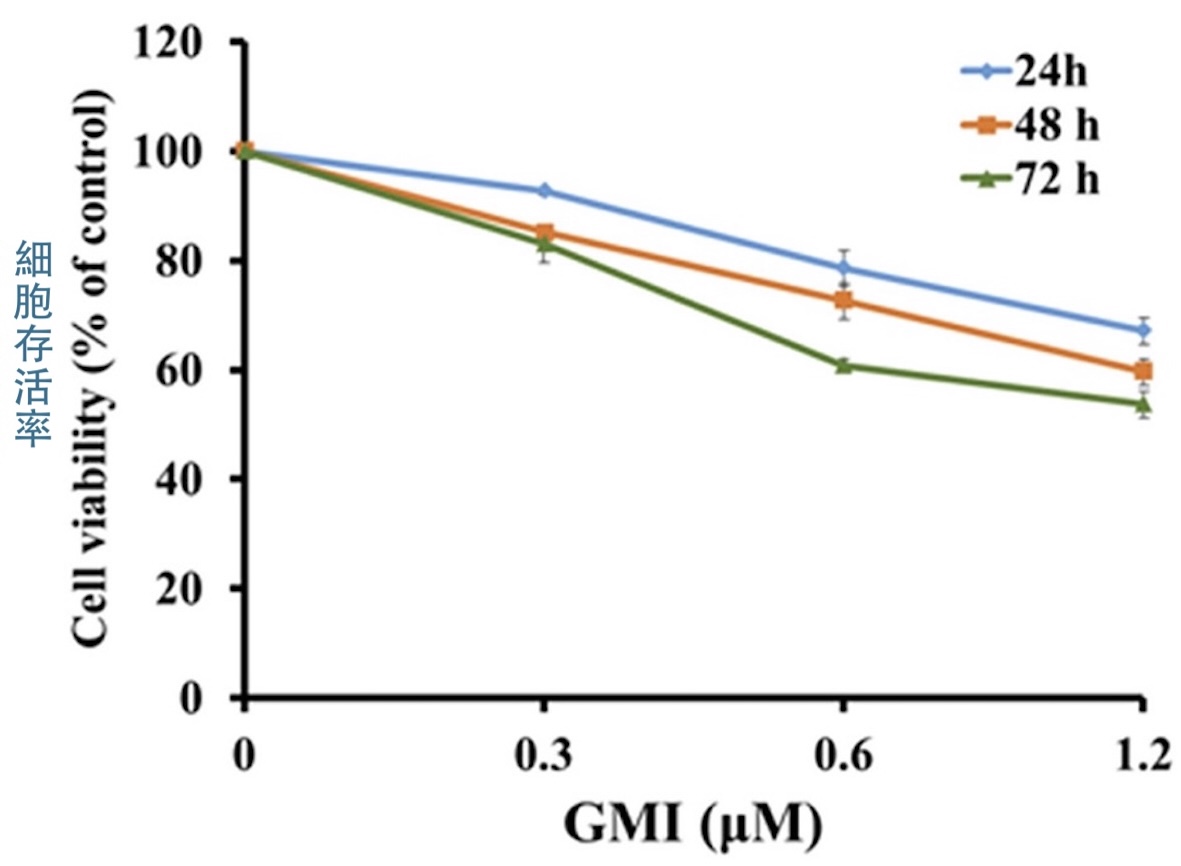
【圖3】GMI能抑制H1975/TR細胞的存活率
(2) GMI能削弱癌幹細胞的勢力
除了透過EGFR突變讓標靶藥失效,癌幹細胞也是抗藥性的一大來源,其在整體癌細胞裡的占比常與抗藥性呈負相關。可是本研究的H1975/TR細胞在跟GMI一起培養12天之後,癌細胞聚集形成的腫瘤球體(tumor spheres)的數量明顯少於沒有GMI處理的組別【圖4】。形成腫瘤球體只有癌幹細胞才做得到,一般癌細胞無此能力,因此實驗結果說明GMI對癌幹細胞也有效——不是能夠阻止一般癌細胞發獲得幹細胞特性,就是可以抑制既有的癌幹細胞增殖。
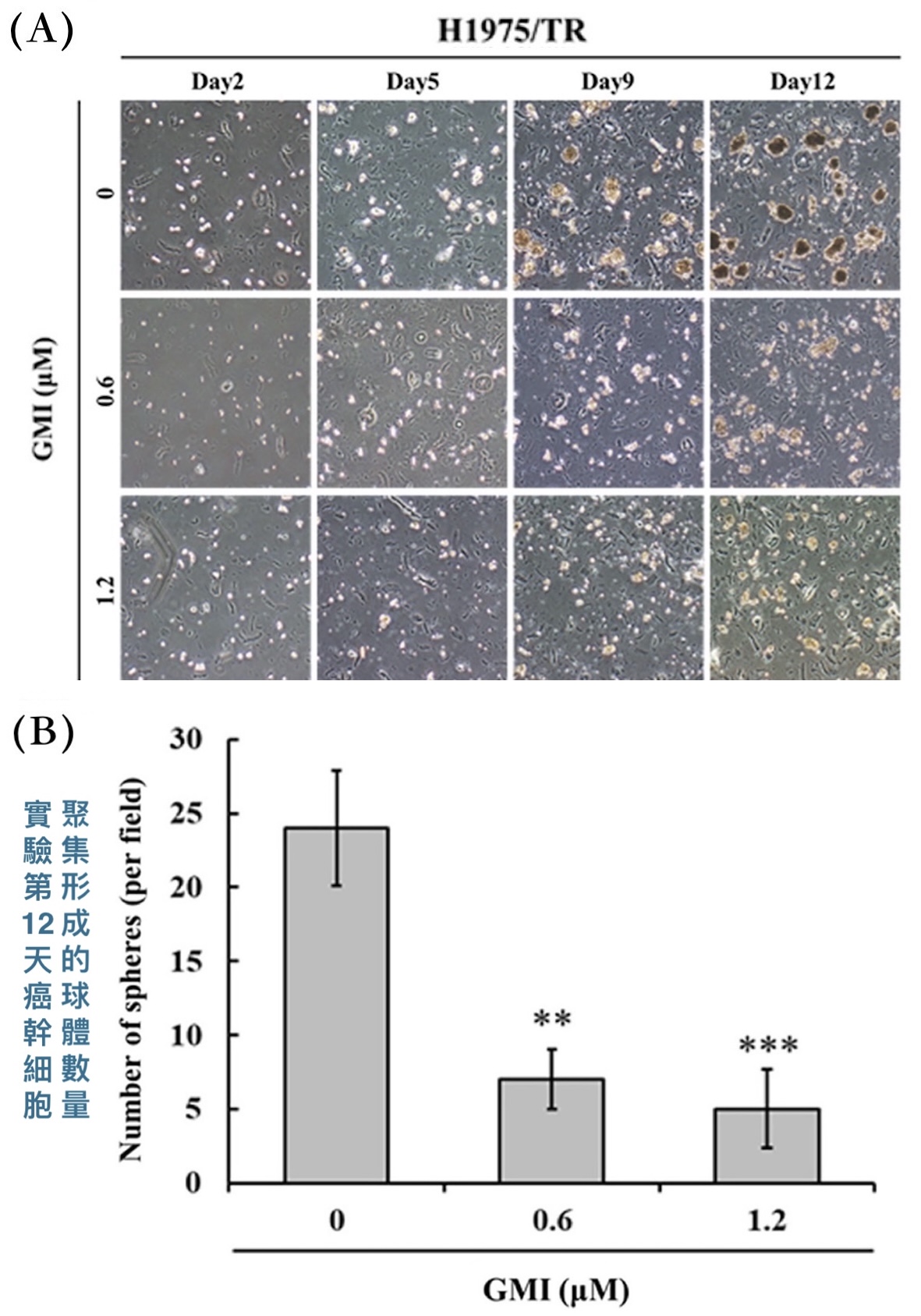
【圖4】GMI能減少H1975/TR形成腫瘤球體(癌幹細胞)的數量
GMI的抗癌機制:不只清除EGFR
也阻止癌細胞透過「整合素」另謀生機
癌症的本質是「細胞生長失控」。對於大量表現EGFR的肺腺癌細胞來說,EGFR就是那個造成失控的開關,因此把EGFR「卡住」,讓它無法啟動細胞增生相關的訊息通路,自然成為標靶藥的設計核心。但也因為標靶藥的藥效跟EGFR的結構息相關,而癌細胞又會靠EGFR突變來掙脫束縛,使得標靶治療宛如一場計畫永遠趕不上變化的結構競賽。
相較之下,GMI化細胞生長失控為可控的做法比較像是斧底抽薪。根據本研究的實驗顯示,GMI不僅會讓突變的EGFR從癌細胞表面消失,還會順勢清除或抑制另一群常被EGFR突變癌細胞拿來另闢蹊徑而大量表達的受體——整合素(integrins)——使癌細胞無法透過這些「備用開關」繼續失控地快速增生、向外遷徒(轉移),甚至發展成癌幹細胞【圖5-7】。
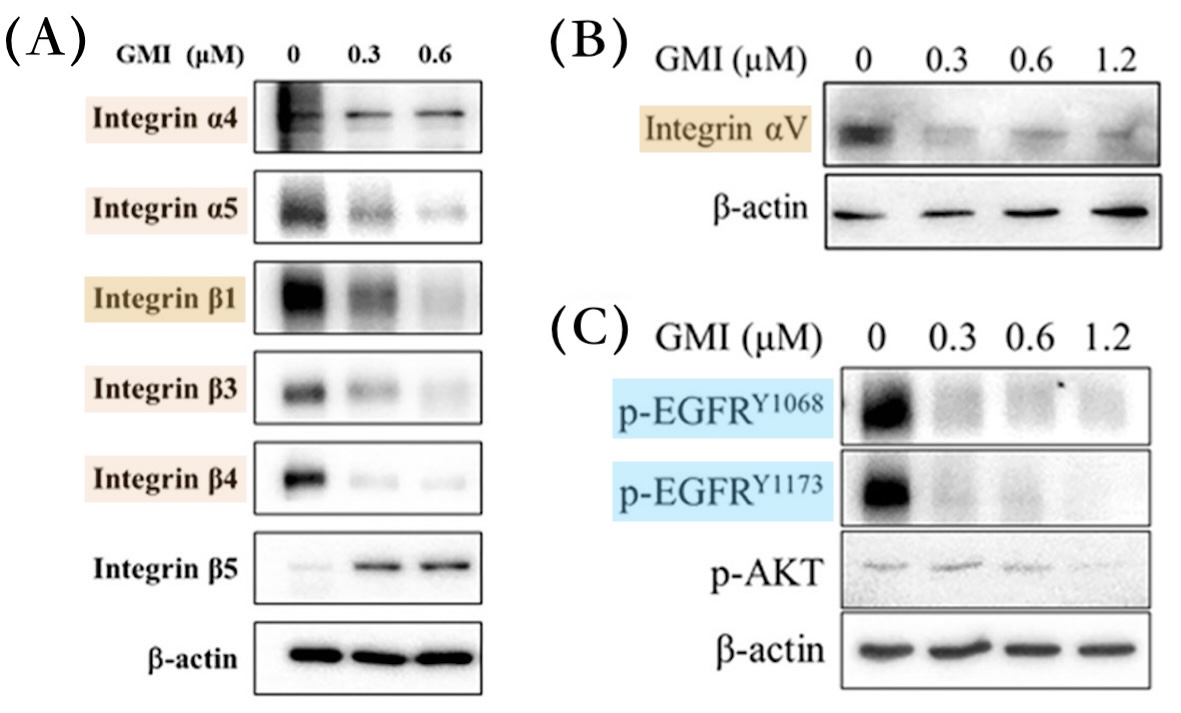
【圖5】GMI可清除或減少H1975細胞的EGFR與整合素
以不同濃度GMI(0.3~1.2 μM)處理H1975細胞:(A) 48小時後整合素α4、α5、β1、β3和β4顯著減少;(B, C) 24小時後整合素αv和突變的EGFR均盡乎消失。
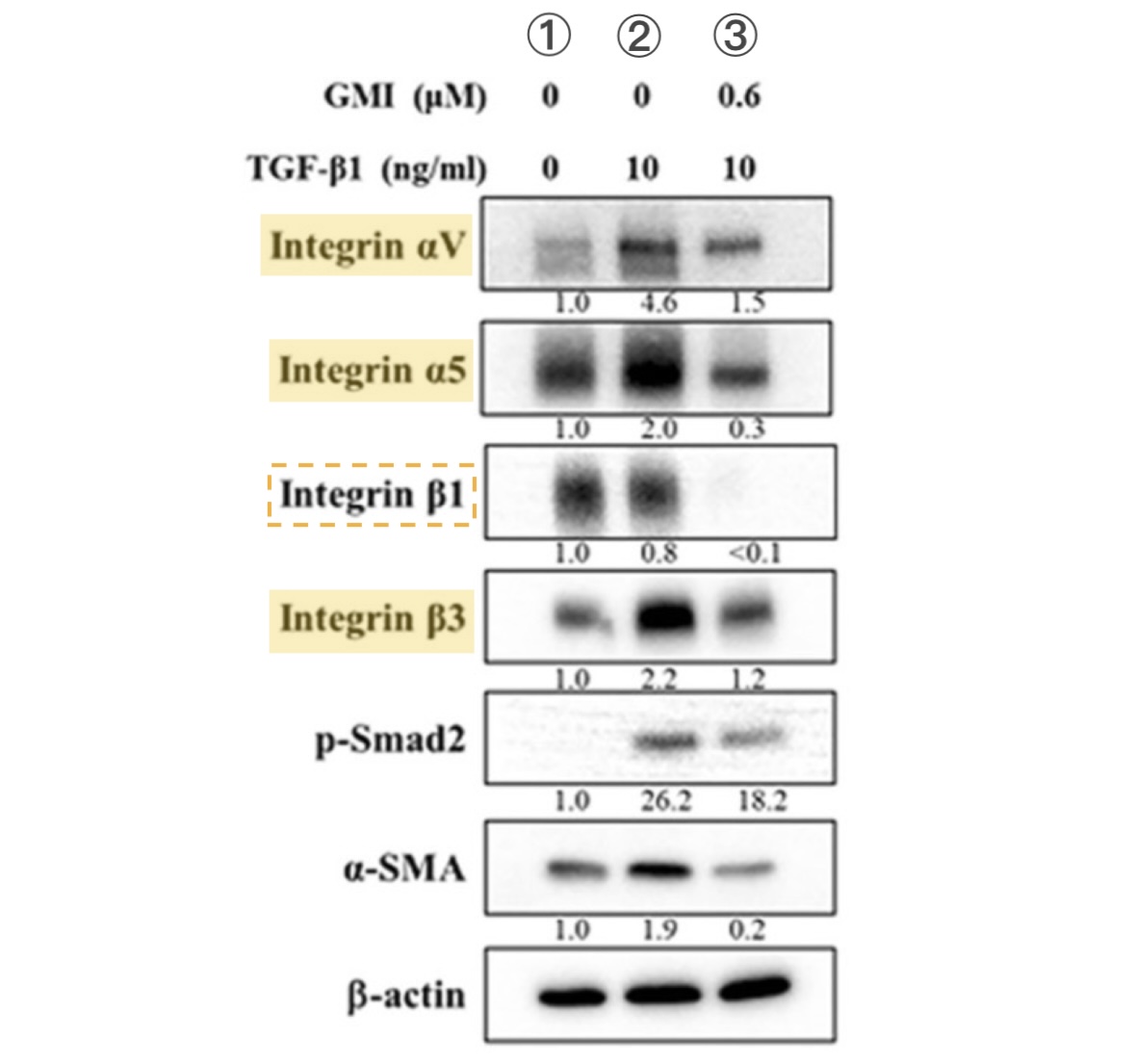
【圖6】TGF-β促使H1975細胞整合素表現量增加的作用可被GMI抑制
有利細胞遷移的轉化生長因子TGF-β會誘導H1975細胞表現更多的整合素αV、α5和β3(① vs. ②),但如果同時有GMI(0.6 μM)存在,這些整合素只會些微幅增加甚至變少,至於僅受TGF-β些微影響的整合素β1則完全偵測不到(③)。
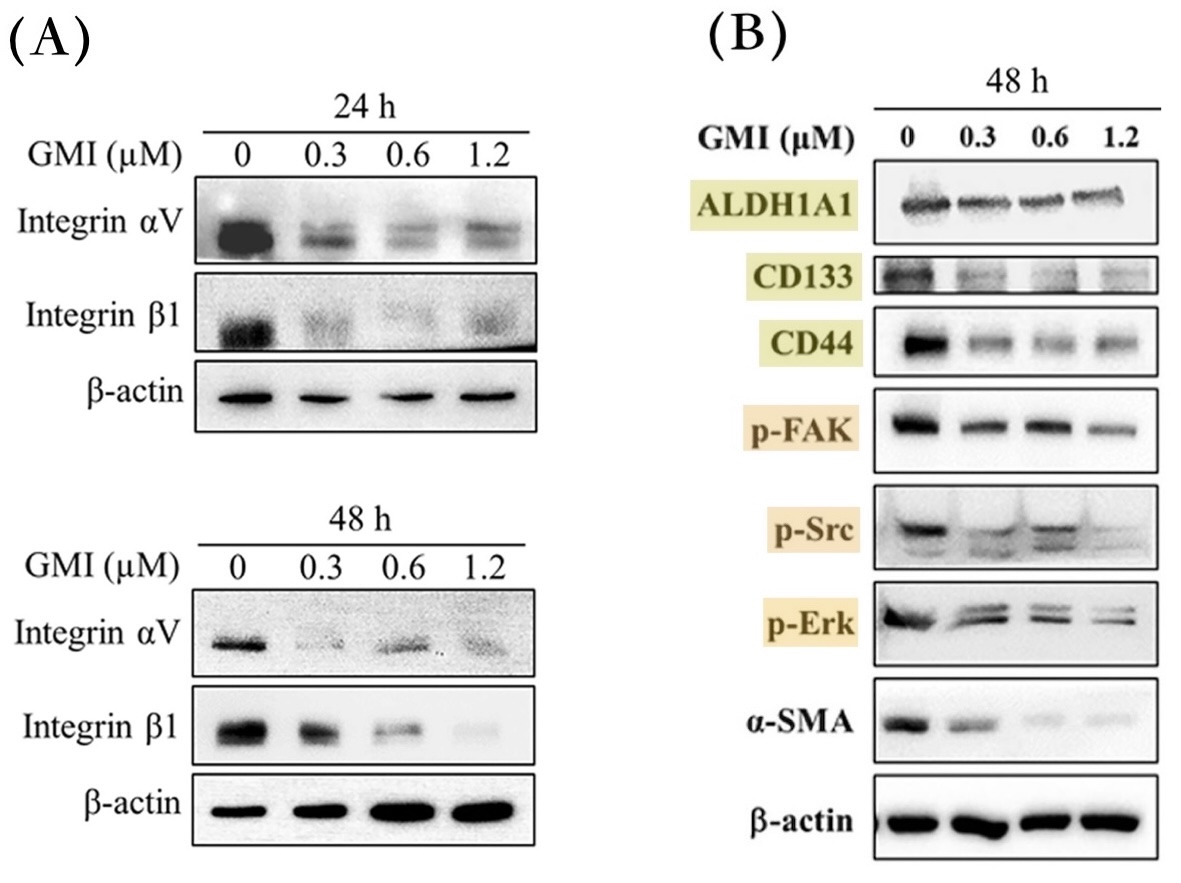
【圖7】GMI可清除或減少H1975/TR細胞的整合素與癌幹細胞
比起H1975細胞,H1975/TR細胞會表現更多的整合素αV和β1,細胞內部被整合素活化的FAK、Src、Erk等分子信號也變得更強(有助細胞遷移和癌幹細胞發展),癌幹細胞特有的分子標記ALDH1A1、CD133、CD44也更顯著(表示有更多的癌幹細胞)。這些指標都會在GMI(0.3~1.2 μM)處理24~48小時之後變少或減弱。
GMI可調控不利癌患生存的整合素αV和β1,
降低其對抗藥性、癌轉移、癌幹性的促進作用
成員眾多、兩兩一組(α型+β型)一起工作的整合素有很多功能,除了可以像鉤子一般幫助細胞穩定附著在周圍組織(細胞外基質ECM),也可以像感應器那樣接收周圍環境的訊息來影響細胞的行為(例如增生或遷移),甚至是細胞遷移時實際負責移動的「腳」。但就如同EGFR的問題一樣,本應受到嚴格調控的整合素卻常被癌細胞過度表現,對癌症治療帶來不利的發展。
(1) 整合素αV和β1 vs. 生存期與癌幹細胞
本研究根據癌症基因資料庫OncoDB的數據進行分析發現,肺腺癌患者比一般人的肺組織有更高的整合素αV基因表現量;癌幹細胞分子標記CD44的基因表現量,與整合素αV、β1的基因表現量都有呈正相關。此外,透過基因表達分析工具GEPIA得到的結果則顯示,整合素αV和β1的mRNA表現量越高,對肺腺癌患者的生存越不利【圖8】。
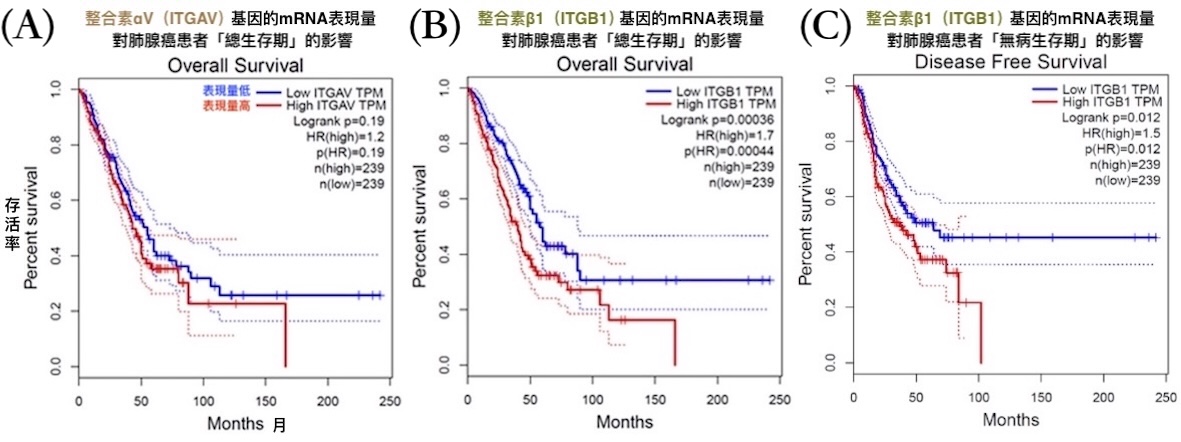
【圖8】整合素αV、β1的基因表現量顯著影響肺腺癌患者的「總生存期」與「無病生存期」
「總生存期」是指從診斷或治療開始到死亡的時間;「無病生存期」是指治療後無癌症復發或病變的時間。mRNA是基因經轉錄作用產生的分子,負責攜帶製造蛋白質(此處指整合素)的訊息,通常mRNA表現量越高,細胞合成該蛋白質的量也越多。
(2) 整合素αV和β1 vs. 抗藥性與遷移性
其他相關研究則證實,當EGFR突變的肺腺癌細胞遭受標靶藥挑戰時,會透過大量表現整合素αV和β1來活化細胞內部的訊息傳遞分子(如FAK、Src等),增強癌細胞的存活能力,使其具備抗藥性【圖9】。此外,肺腺癌細胞的遷移能力也跟整合素αV、β1的表現量,以及被整合素活化的FAK活性呈正相關。
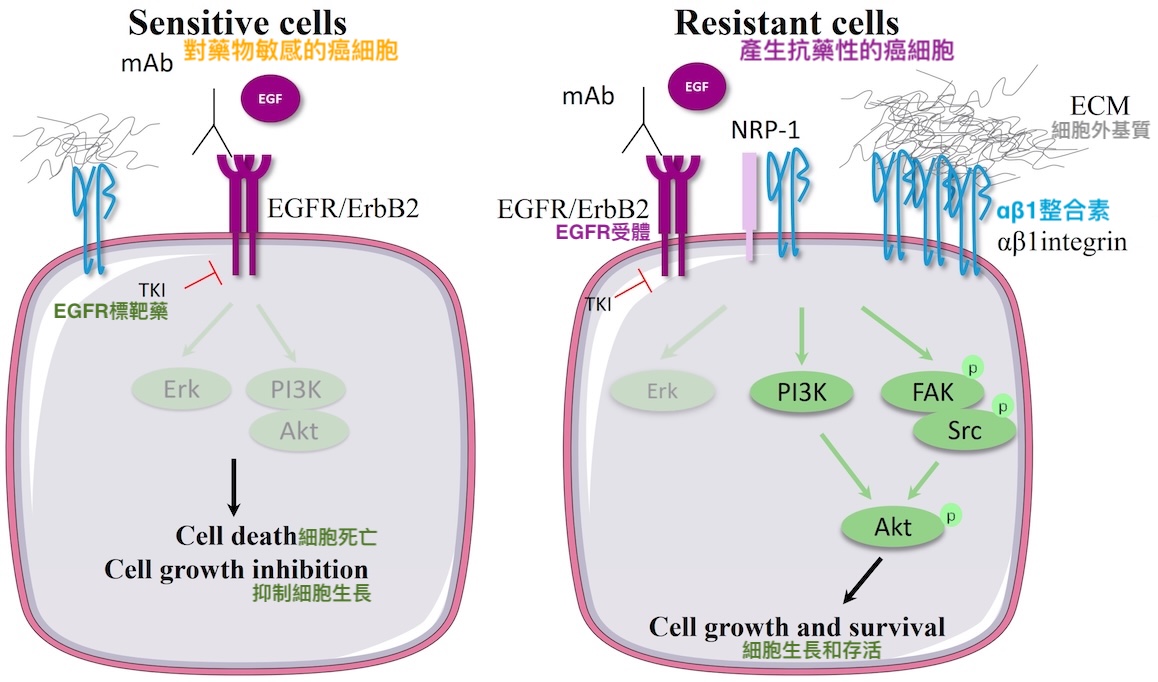
【圖9】EGFR突變型癌細胞藉由過度表現整合素β1對EGFR標靶藥產生抗藥性示意圖
EGFR標靶藥都是透過跟EGFR的酪胺酸激酶結合來發揮作用,故統稱為「酪胺酸激酶抑制劑」(tyrosine kinase inhibitor,簡稱TKI)。(圖片來源:https://www.mdpi.com/2072-6694/11/5/692)
(3) GMI vs. 整合素αV和β1
總的來說,整合素αV和β1不只有助EGFR突變的肺腺癌細胞產生抗藥性、增強遷移性、獲得癌幹性,臨床上還不利肺腺癌患者生存。
而在本研究裡的EGFR雙突變人類肺腺癌細胞,不論是對前兩代標靶藥有抗藥性的H1975細胞,還是在TGFβ誘導下增強遷移性的H1975細胞,或是對第三代標靶藥有抗藥性且具有顯著幹細胞特性的H1975/TR,都能因為GMI介入而減少細胞表面的整合素αV、β1【圖5-7】及細胞內FAK、Src、Erk分子的活性【圖7】。
此作用機制【圖9】對於GMI為什麼能抑制EGFR雙突變肺腺癌細胞的腫瘤生長、遷移能力、生存能力和癌幹細胞特性【圖1-4】提供了部分解釋——因為GMI能讓此路不通(被EGFR標靶藥阻斷)而山不轉路轉(大量表現整合素)的癌細胞,連替代道路(整合素調控的訊息傳遞路徑)也開通不了,或是即使開通也無法一路通暢到底(提高癌細胞獲得遷移能力和幹細胞特性的困難度)。
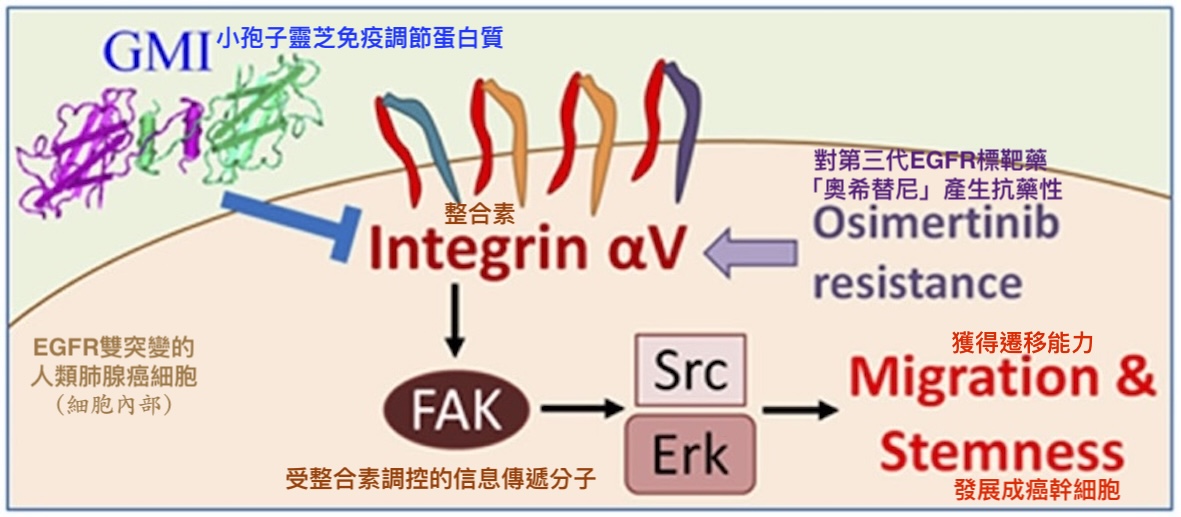
【圖10】GMI抗癌機制示意圖——以調控整合素αV為例
GMI可阻斷整合素αV啟動的訊息傳遞通路(Integrin/FAK/Scr/Erk),抑制對奧希替尼有抗藥性的EGFR雙突變人類肺腺癌細胞獲得遷移能力與幹細胞特性。
清除過量的整合素與EGFR
GMI讓癌細胞從失控回到可控
本研究揭示GMI可以經由調控整合素來抑制EGFR雙突變人類肺腺癌細胞的全新作用,跟陽明交通大學林東毅教授團隊2023年發表的研究成果頗有異曲同工之妙。該研究在小鼠肺腺癌細胞LLC-1(對第一代EGFR標靶藥有抗藥性)的動物實驗即觀察到,以腹腔注射方式進入體內的GMI不只抑制腫瘤生長,也能大幅減少腫瘤組織裡多種有利癌轉移的整合素(詳見相關報導:GMI抗肺腺癌轉移的分子機制新發現:讓癌細胞綁手綁腳,使其跑不了和尚也建不了廟)。
此外,本研究觀察到GMI可以讓突變型EGFR從人類肺腺癌細胞上消失的作用,也與陽明交通大學林東毅教授團隊2023年發表的研究成果一致,其箇中原理在林的論文亦有詳細闡述(詳見相關報導:GMI抑制EGFR陽性肺腺癌腫瘤的重要機制——迫使癌細胞吞掉自己的EGFR)。
如前所述,EGFR和整合素都是正常細胞會有的基本配備,差別在正常細胞只在需要時才把它們拿出來適度使用,用完之後就被細胞資源回收再利用;而癌細胞則是毫無限度地將它們大量表現、拼命使用,用到連旁邊的正常細胞也跟著「變壞」。從現有的研究結果看來,源自靈芝家族、結構渾然天成的GMI【圖11】顯然是讓癌細胞自廢武功,讓它們從失控的變態回歸到可控的常態。當失序的細胞少了,健康新秩序的重建應該也就不遠了。
【圖11】小孢子靈芝免疫調節蛋白GMI的化學結構
由111個胺基酸組成的小孢子靈芝免疫調節蛋白GMI是一個純的蛋白質。圖為GMI在蛋白質資料庫PDB登錄的立體結構,代號3KCW。
〔資料來源〕Yu-Ting Kang, et al. Integrin αV Inhibition by GMI, a Ganoderma Microsporum Immunomodulatory Protein, Abolish Stemness and Migration in EGFR-Mutated Lung Cancer Cells Resistant to Osimertinib. Environ Toxicol. 2024 Dec;39(12):5238-5249. doi: 10.1002/tox.24399.
〔附圖文獻〕Cruz da Silva E, et al. Role of Integrins in Resistance to Therapies Targeting Growth Factor Receptors in Cancer. Cancers. 2019; 11(5):692.